What you need to know about COVID-19 testing
Posted on: Friday 19 June 2020
Author: Robert Holland FFPM and John Bagshaw
Foreword
Professor Tim Higenbottam, FPM president
Testing for SARS-CoV-2, the causative agent of COVID-19, has been the subject of daily reporting in the UK and around the world for many weeks. There has been a focus on reporting of the ‘ramping-up’ of testing numbers, together with initial excitement and then disappointment regarding so-called ‘finger prick’ antibody tests. Now seems a good moment to reflect upon the actual purposes of testing, and to discuss the science behind the various existing methodologies, to focus on the questions they can really answer, and their limitations.
Professional membership bodies and medical royal colleges are stepping up and supporting and influencing the national programme. The Royal College of Pathologists have recently published ‘Testing for the Future – A National Strategy’, which sets out a vision for a future strategy for SARS-CoV-2. Faculty of Pharmaceutical Medicine members are also contributing, especially those who work in the field of development of in vitro devices. There is an extensive and effective in vitro diagnostics industry in the UK, which employs a considerable number of highly qualified individuals and plays an important role in the UK’s vibrant life sciences sector. For this comprehensive and authoritative blog article, we are pleased to be joined by an author from the British In Vitro Diagnostics Association (BIVDA).
From figure 1, we can see the daily number of patients with a confirmed positive test for SARS-CoV-2, using reverse transcription polymerase chain reaction (RTPCR), during the UK epidemic. This epidemiological data provides some understanding about the rapid spread of the illness and the impact of the social ‘lock down’ in gaining control. It offers one measure to demonstrate to us that the initial epidemic is in decline.
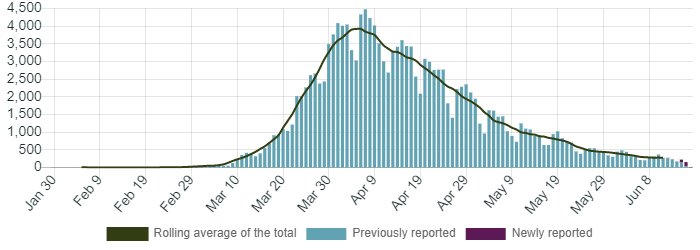
Figure 1. Daily number of lab-confirmed cases in England by specimen date. Total confirmed cases showing those previously reported and newly added cases separately. New cases are attributed to the day the specimen was taken. [Image reproduced with permission from https://coronavirus.data.gov.uk/]
The capacity for testing is a vital ingredient to the proposed “Test and Trace” strategy that is being initiated by Government and is being directed by Baroness Dido Harding. Although the epidemic overall is in decline, we have to anticipate local outbreaks. Test and Trace will be vital in the control of such outbreaks. It aims to achieve two main goals:
- To ensure that anyone who develops symptoms of coronavirus (COVID-19) can quickly be tested to find out if they have the virus, (including targeted asymptomatic testing of NHS and social care staff and care home residents)
- To trace close recent contacts of anyone who tests positive for coronavirus and, if necessary, notify them that they must self-isolate at home to help stop the spread of the virus
Not only is an increased capacity of testing needed but an integrated nationwide organisation needs to be in place that can both test and trace, but also communicate quickly. In order to keep the value of R well below 1.0, we need to ensure testing on an extraordinary scale and effective tracing, coupled with the support of a trusting population that will follow the advice on self-isolation.
Beyond the epidemiology and the control of future local outbreaks of infection, testing for SARS-CoV-2 infection also requires an understanding of whether people become immune to this virus after an infection. This requires significant clinical development, often forgotten in a crisis, but described in detail in the following review of testing.
The following article has been prepared by Robert L Holland FFPM & John Bagshaw, Interim Chief Operating Officer of the British In Vitro Diagnostics Association (BIVDA).
It is provided for information and does not constitute advice or represent official FPM views or policy.
How to cite:
Holland and Bagshaw (2020), ‘What you need to know about COVID-19 testing’, Faculty of Pharmaceutical Medicine blog, 19 June 2020. Available at: https://www.fpm.org.uk/blog/what-you-need-to-know-about-covid-19-testing/ (Accessed: <date>).
Diagnostic test development and evaluation
In the pharmaceutical industry we are often somewhat blind to the realities of what constitutes the evaluation of an in-vitro diagnostic test – and how those evaluations are described, the so-called “performance characteristics”. It is worth remembering that the performance characteristics of a test need to be thought of in three ways:
1. Analytical Validation
Does the device actually work as described and has it been manufactured properly? Are the materials and reagents consistent and reliable from batch to batch, and from individual device to device? Do different users get the same results with same samples? In what environments does the device actually work? Does the user need special training? If the device is measuring the quantities of an analyte, what is the lowest amount of that analyte that the device can reliably detect? Are there any other materials potentially in a sample which might distort the result? (It is relevant here to remember that SARS-CoV-2 resembles other coronaviruses, both at the RNA level and in terms of some of the proteins present in the capsid or induced in the host). What kinds of samples (transported under what kinds of conditions) provide a reliable result? Most of the very large number of different Covid-19 tests available or in development have had some sort of analytical validation – but this has often not been to the standards one might anticipate. It also needs to be understood that tests described as CE-marked may have had no third-party scrutiny and that FDA “emergency use authorisation” implies a very limited degree of independent evaluation.
2. Clinical Validation
This is usually expressed in terms of sensitivity and specificity and sometimes combined as accuracy. Sensitivity is the proportion of all true positive samples called positive by the test. Specificity is the proportion of all true negative samples called negative by the test. Many tests have been described as having extremely high levels of sensitivity and specificity (up to 99% or so for each) – but it needs to be understood that sensitivities and specificities are comparatives not absolutes and a key matters often not described in the literature accompanying a test is “what were the samples used as “True Positives” and “True Negatives”” and “how was that status determined”? Without knowing the nature of the validation samples, sensitivities and specificities are difficult to interpret. Examples of this were the early COVID-19 antibody tests, where the validation samples were from patients who had experienced a severe illness, the samples were taken at the height of the patient’s IgG response and the samples had already been shown to contain high levels of antibody through other serological techniques. Although the data are not yet completely clear, it appears that a small minority of patients may not develop an antibody response and those that do, evolve the antibodies over time, generating IgM around ten days post infection and IgG and IgA a few days later. This means that the sensitivities and specificities quoted could be quite misleading for the diagnosis of active disease.
3. Clinical Utility
This is a critical matter but is rarely fully understood and even more rarely fully explored for commercially available in vitro diagnostics. This concerns an evaluation of the clinical usefulness of a result from the test (and is tied to the list of questions below asking what the test might be used to determine). Unfortunately, since our understanding of COVID- 19 is still at an early stage, the meaningfulness of a result can be a matter of debate even though the result might be both accurate and reliable. Thus, we can detect specific SARS-Cov2 RNA from throat and nasopharyngeal swabs (if taken properly), and with less sensitivity from saliva. Detection certainly means the subject has been exposed to the virus. But, absent signs and symptoms, will they develop the illness or not? Are they infectious? Today we must assume they could be infectious but since we don’t fully understand how the virus is transmitted, let alone how much viral exposure is required for infection, there is no definitive answer to these questions. Especially if a result is to be used for screening for a disease, or for epidemiology, it is critical to have information on the true prevalence of positivity in the population tested, and the population size, in order to assess the true value of the test (its positive and negative predictive value). A test which has a sensitivity and specificity of 90% may be useful when there is a high prevalence and the object is to capture most of the true positives. If 50 of a population of 100 are positive, this test will detect 45 of them and wrongly indicate 5 as negative, but will also give 10 false positives. Its Positive Predictive Value (PPV) is therefore 45/55 = 82%. If, however only 10 in 100 are true positives, it will detect 9 true positives, but still 10 false positives, so its PPV = 9/19 = 47%. Now that it appears that the prevalence of COVID-19 is rather low and falling (reported as 0.1% by the Office of National Statistics on 5th June) the issue of false positives may become a real problem. For antibody testing to determine whether an individual has recovered from an infection and is therefore immune (assuming that is a given) the highest possible specificity is required for safety. For diagnosis, say using a PCR test on a swab to detect a virus, the priority is sensitivity, and a slight compromise may be made on specificity. This applies, for example to HIV diagnostic tests, where 100% sensitivity is the aim, and confirmation of a positive test result is always required before reporting the result.
Who should be tested and why?
The perceived reasons for testing for COVID-19 (the clinical utilities of the test) vary according to who is requesting the test but can be roughly summarised as follows:
- To confirm a diagnosis of COVID-19
- by a treating physician, to ensure that the patient receives the most appropriate treatment for their actual illness (i.e. is it COVID-19 or is it something else possibly requiring an alternative therapy)
- by the public health system
- to isolate a potentially infectious person
- to trace contacts and to isolate the contacts if infected
- by the individual – for reassurance or to confirm their need to isolate
- To establish that an individual is not infectious
- by the work-place or other controlled circumstance where individuals might gather, for communal reassurance that the gathering is safe and effectively quarantined
- by the individual to give reassurance for freedom of movement and travel
- To understand the epidemiology of COVID-19
- by professional epidemiologists, health care providers, and the government
- to understand the current prevalence of actively infectious individuals and clusters of infection and therefore to monitor the evolution of the epidemic and intervene as appropriate
- to understand who has already been infected, when and how they were infected and therefore to predict the future evolution of the epidemic
- by the life sciences industry and academia
- to design clinical trials of drugs, vaccines and diagnostics effectively
- by professional epidemiologists, health care providers, and the government
- To understand immunity to COVID-19
- by epidemiologists and public health specialists, to inform models of the spread of the epidemic
- by academics and the life sciences industry
- to develop effective and safe vaccines
- to understand the complex and varied immune response to the virus.
It is not feasible that one test can meet all these various requirements. It also seems obvious that a focus on only the numbers of tests (and not even the numbers of individuals who receive a result) is a distraction from the real and urgent requirement to meet all of the above needs with a variety of effective tests, with performance characteristics appropriate to the question that the test is being used to address. Until now the focus has been on tests to detect the presence of COVID-19 for purposes 1, 2 and 3 above, and negative test results have been accepted as evidence that the individual is not infected and has not been infected within the previous three weeks or so, i.e. is extremely unlikely to transmit the disease. No large-scale immunity studies have yet been performed in the UK.
Types of tests currently in use and in development for COVID-19
Turning to the test themselves, those currently available and those in development. How do they work and what questions should they be used to address? Tests fall into two basic categories: those to detect the virus itself (either viral RNA or viral protein); and those to detect an immune response to the virus.
1. Detection of the virus itself or of viral materials
a. Detection of viral RNA by RT-PCR.
The SARS-CoV-2 genetic sequence was revealed by researchers from China around 10-12 January 2020 (1, 2) and deposited in Genbank (accession MN908947). This has allowed the very rapid design of primers and other reagents for Reverse Transcription Polymerase Chain Reaction (RT-PCR) methodologies to detect SARS-CoV-2 viral RNA. Many of the tests can simultaneously distinguish SARS-CoV-2 from other similar viruses such as SARS and MERS as well as the more common corona viruses endemic in humans 229E, NL63, OC43, and HKU1 and sometimes, various influenza viruses. RT-PCR is a well-established, and very commonly used technology for the diagnosis (and monitoring e.g. of HIV) of viral infections and is widely available in clinical diagnostic laboratories.
As of 7 June 2020, there were at least 146 commercially available in-vitro diagnostic (IVD) PCR kits (combinations of consumables and reagents for PCR machines in clinical laboratories) – an increase of 38 over the previous month (3). These kits are generally based around the detection of one or two conserved genes: RdRP – the RNA dependent RNA polymerase gene found within the orf1ab region; the E (Envelope protein) gene; and the N (nucleocapsid protein) gene.
As would be anticipated for such a well-established technology, all but eight of these manufacturers claim some sort of regulatory approval in Europe (CE-mark) or the USA (EUA) for their tests; however, the majority of these “approvals” are actually Emergency Use Authorisations (EUA) issued by the FDA or occasionally the US CDC. Other tests (around 50) claim CE-mark, but it should be remembered that this will generally be by self-certification. The implications of this are that any laboratory using these tests will need to establish for themselves the functionality of the kits on their devices and maintain rigorous quality control procedures including the processing of standard positive and negative samples on a very regular basis. It also means that switching between kits may not be possible (because the kits will have been analytically validated for specific PCR machines), or at a minimum a switch will require extensive revalidation of the laboratory processes and work-flows.
Key questions for any diagnostic test are what sample type (tissue or fluid) should be collected, and when and how to collect those samples. In cohorts of symptomatic patients, viral nucleic acid is reported to be detected in respiratory samples (nasopharyngeal and/or oropharyngeal swabs, aspirates, sputum) at symptom onset and for up to two to four weeks (occasionally longer) (4, 5). Initial viral loads in posterior nasopharyngeal specimens are reported to be high, but somewhat lower in saliva (and not always detectable); load is reported to decline slowly in recovering patients and to be somewhat higher at presentation in more severe patients and older patients. Viral nucleic acid was detected in stool or rectal swabs in 59% of patients and in serum in 41%.
For obvious reasons, most of the literature describing studies of PCR based viral nucleic acid detection have concentrated upon symptomatic patients (in many countries, the presence of symptoms has been, at least for some periods, a pre-requisite for testing). However, there are reports of asymptomatic carriers testing positive (6, 7, 8, 9). These individuals were typically contacts of known infected patients. In these reports many (but not all) of the subjects became symptomatic; in the pre-symptomatic individuals viral nucleic acid was detected on average 3 days before the emergence of symptoms.
A key matter is the turnaround time between obtaining a sample and the delivery of results to the patient and their physician. Although RT-PCR is a relatively quick technology (running the test manually from sample to result can take 3-4 hours, and automated systems can reduce this time) delays can occur due to logistical issues with sample transport, results review, and return of the results. Other delays occur due to machine down-times, and reagent supply issues. Queues of samples can build up at centralised testing laboratories. It ought to be possible when testing is performed in a local laboratory for results to be returned within less than 24 hours, but in territories where testing has been centralised, there have been reports of considerable delays.
Returning to the list of clinical utility questions above, the following conclusions regarding RT-PCR seem sensible:
- RT-PCR has the characteristics of a very good diagnostic test; it has excellent sensitivity and specificity (at least with respiratory samples) and meets the needs of all parties when the results are returned quickly.
- The situation regarding infectivity is less clear. A positive test probably means someone is potentially infectious although viral nucleic acid is detectable long after the symptomatic period for many patients. More research will be needed to confirm whether current guideline in the UK, for example, of 7 days self-isolation is sufficient. A negative test means only that the individual is unlikely to be infectious at the timepoint that the sample was taken. Given the reports that asymptomatic individuals exist and pre-symptomatic individuals harbour virus for three or more days before showing symptoms, rather frequent testing will be required (perhaps twice weekly) to guarantee that an individual is not infectious. Again, this guarantee of non-infectivity requires rapid turn-around of the results.
- RT-PCR is probably excellent for its use to evaluate the current prevalence of disease and to locate infection hot-spots.
- It is not useful as a method to determine how many people have been previously infected nor who might be immune.
b. Detection of viral nucleic acid by methods other than RT-PCR
The need for a more rapid turnaround time of results and perhaps for greater sensitivity in samples such as saliva has driven the invention and development of alternatives to classical RT-PCR.
Four such EUA approved tests with substantially reduced turnaround times use isothermal amplification technology. One example is the Abbott ID-Now near patient device, which detects the RdRp gene in less than 15 minutes directly from throat and nasopharyngeal swabs. This technology seems potentially extremely useful as a means to demonstrate non-infectivity, and possibly useful in situations where rapid confirmation of a clinical diagnosis is required. The Abbott kit and device seem slightly less sensitive in situations of reduced viral quantities through dilution in, for example, viral transport media. Other comparable tests are in development and are expected to become available in the future. Another very interesting technology for the rapid detection of viral nucleic acid which has become commercially available in the last month uses a CRISPR technique to enzymatically detect and signal the presence of the nucleic acid. This technique is reported to provide results within one hour from nasopharyngeal and throat swabs, and other respiratory samples (10).
Some rapid PCR systems run panels of tests simultaneously, e.g. for lower respiratory infections, and can identify which causative agent is present, rather than giving only a yes/no answer for a single test, and in approximately 1 hour. These systems do not yet include COVID-19 in FDA, EUA or CE-marked panels but it is only a matter of time.
Droplet digital PCR is a technology to improve the sensitivity in samples with relatively fewer copies of viral nuclei acid and reduce the rates of false negatives. One such test is available (BioRad, FDA EUA) and others are in development or awaiting approval. The clinical utility of this kind of test is not yet completely clear.
c. Detection of viral material which is not nucleic acid.
On May 9th, the FDA granted emergency use authorisation for a very exciting alternative to nucleic acid testing for the SARS-CoV-2 virus – a device and reagents that qualitatively detect the presence of the nucleocapsid protein. The Sofia 2 SARS Antigen FIA is lateral flow immunofluorescent sandwich assay to be used with the Sofia 2 Fluorescent Immunoassay Analyzer from Quidel (11). This is a near patient or point of care test which needs to be carried out in suitable facilities with trained staff, but which can provide results in a matter of minutes from throat and nasopharyngeal swab samples. The precise performance characteristics of this IVD are not yet clear, but the manufacturer warns that similar antigen tests are known to be less sensitive than nucleic acid tests. In the last few days, a similar test has gained a CE-mark in Europe (Maripoc from ArcDia).
Antigen tests in lateral flow devices are also becoming available and may well have a place in circumstances such as cruise ships and offshore establishments, e.g. oil rigs, where a rapid test for current infection is highly valued. Such tests for Influenza and Norovirus have been used for some years, and, although less sensitive than PCR techniques, can provide a fast confirmation to help limit outbreaks in these confined and remote locations.
2. Antibody tests and Serology
Antibody tests have generated significant controversy. Governments and healthcare providers initially purchased millions of IVD kits and then discovered them to be unsatisfactory (at least for the purposes for which they were bought), because of high numbers of apparently false negatives and false positives. However, now that we are beginning to better understand the time course of the antibody response to COVID-19 and the risks of cross-reactivity with antibodies to other corona viruses (5, 12), it seems that antibody testing kits are finding their clinical utility as epidemiological tools to detect previous infection by COVID-19. They can possibly be used to show current individual and community levels of immunity, and perhaps to identify individuals who have seroconverted and from whom therapeutic antibodies might be obtained. Indeed, on 6th June the UK Government announced that almost 200,000 antibody tests had been performed in the previous week in the four centralised laboratories established for this purpose. It has also been suggested that an antibody test could be useful in ambiguous cases of clinically diagnosed COVID-19 (perhaps on the basis of abnormal CT scans) where RT-PCR is negative but there is a suspicion that this might be a false negative.
The SARS-CoV-2 virus contains four structural proteins: the spike [S], membrane [M], envelope [E], and nucleocapsid [N] proteins. As is now well-known, the Spike (S) protein interacts with the Angiotensin Converting Enzyme 2 expressed on the surface of many human cells to enable viral ingress. The S protein is immunogenic and the receptor binding domain of the S protein is the target of neutralising antibodies that have been reported from day 9 onwards in infected patients (13, 14). Antibodies to the N protein are also detected in patients with COVID-19 (15) although there can be issues of cross-reactivity with other coronaviruses.
Traditional serology for the detection of neutralising antibodies would use replication competent virus in cell culture, might take several days, and would need to be conducted in a biosafety level 3 laboratory environment. Therefore, enzyme linked immunosorbent assays (ELISA) and immune fluorescence assays (IFA) have been developed using recombinant S (S1 and S2) (13) and N proteins as antigen to capture and detect antibodies. Other techniques (e.g. chemiluminescence methods) also use recombinant viral protein to bind antigen and generate a signal. Some of these tests (e.g. ELISA and IFA) can then be configured as hand-held devices, others are more suited to laboratory detection of antibody (e.g. electro-chemiluminescence) from conventional serum and blood samples, as has been deployed in the UK.
A range of reports are now available describing the evolution of an antibody response to SARS-CoV-2 infection. Essentially, IgM can be detected in about 50% of patients within five days of symptom onset rising to 80% or so at around 10 days; IgG and IgA rise a few days later. Tests which combine detection of IgM and IgG appear to have 90+% sensitivity for known cases of COVID-19 (5, 16, 17, 18, 19), and perhaps even higher at 10-14 days when antibodies against both the S and N proteins are sought and total antibodies measured. It has even been reported that a combined IgM and IgG may be more sensitive in the detection of SARS Cov2 than RT-PCR (e.g. ref 21). However, the rate of false negative testing for viral nucleic acid in this cohort of clinically diagnosed patients seems high and hard to explain.
As of 7 June 2020 there are 38 IVD kits and systems for the detection of antibody to SARS-CoV-2 with some sort of regulatory approval (up from 28 a month previously): 21 CE-marked, 10 under FDA EUA regulations and 7 with both approvals (3). There are at least four more in development with a status of “EUA submission pending” – a marked reduction in number from a month earlier. The difference in numbers in comparison with the PCR tests is striking and probably reflects the general view that antibody tests are unsuitable for diagnosis.
It is worth noting that the current view of Public Health England (7 June 2020) is that antibody tests, formatted as a rapid point of care test or as a finger prick test, are not recommended for use. On the other hand, mass antibody testing remains as pillar 3 in HM Government’s strategy for testing and will likely be critical in the medium term management of the UK’s cautious emergence from “lockdown” as a vital tool to monitor population exposure and likely immunity (see ref 21).
The MHRA have produced specifications for antibody self-tests and for antibody point of care tests, and are evaluating current products and new ones as they become available, in order to find devices that can be useful in a mass testing strategy or for specific purposes. These require a minimum 98% sensitivity and 98% specificity, and, for self-tests, simplicity and reliability of operation. There is also a specification for an ‘end to end’ testing system, comprising sampling devices that can be sent to individuals for self-testing and either sent back for testing or tested on the spot. It may be that the use of rapid point of care or self-tests for antibody will be approved with suitably high performing devices and with the proviso that laboratory based antibody tests of an even higher performance specification are used to confirm any positive results.
Conclusion
The COVID-19 pandemic has demonstrated to a wide audience the critical importance of in vitro diagnostics to healthcare practice in general and public health in particular. If there are any benefits to be drawn from the response to the pandemic, one might be recognition and appreciation of the dynamism and skill of those who invent, manufacture, evaluate and deploy diagnostics; another might be that this effort serves as a catalyst to boost the academic and life sciences Industry investments in the field.
References
- https://www.who.int/csr/don/12-january-2020-novel-coronavirus-china/en/
- http://virological.org/t/novel-2019-coronavirus-genome/319
- https://www.360dx.com/coronavirus-test-tracker-launched-covid-19-tests
- Zheng S et al BMJ. 2020; 369: m1443. Published online 2020 Apr 21. doi: 10.1136/bmj.m1443: 10.1136/bmj.m1443
- To K K-W et al Lancet Infect Dis 2020; 20: 565–74
- Meng H et al https://doi.org/10.1016/j.jinf.2020.04.004
- Wang Y et al The Journal of Infectious Diseases 2020;XX:1–5
- Lai C-C et al https://doi.org/10.1016/j.jmii.2020.02.012
- Zhou J et al https://doi.org/10.1016/j.jinf.2020.03.028
- https://sherlock.bio/sherlock-biosciences-receives-fda-emergency-use-authorization-for-crispr-sars-cov-2-rapid-diagnostic/
- https://www.360dx.com/regulatory-news-fda-approvals/quidel-receives-first-fda-emergency-use-authorization-rapid-poc#.XrgoyGhKiyJ
- Laurenco J et al https://doi.org/10.1101/2020.03.24.20042291
- Amanat F et al https://doi.org/10.1101/2020.03.17.20037713
- Haveri A et al Euro Surveill. 2020;25(11):pii=2000266. https://doi.org/10.2807/1560-7917.ES.2020.25.11.200026
- Guo L et al Clin Infect Dis. 2020 Mar 21. pii: ciaa310. doi: 10.1093/cid/ciaa310
- Zhang, W. et al. Emerging Microbes Infect. 2020, 9 (1), 386−389.
- Xiang, J. et al. medRxiv, March 1, 2020. DOI: 10.1101/2020.02.27.20028787
- Cai, X. et al. medRxiv, February 25, 2020. DOI: 10.1101/2020.02.22.20026617
- Lou, B et al medRxiv preprint doi: https://doi.org/10.1101/2020.03.23.20041707
- Liu, L et al medRxiv preprint doi: https://doi.org/10.1101/2020.03.06.20031856
- https://www.abpi.org.uk/media/8206/update-on-the-national-testing-programme-and-covid-19-surveillance-slides-for-distribution.pdf