COVID-19 Vaccine and Antiviral Drug Development
Posted on: Wednesday 8 April 2020
Author: Author: Dr Penny Ward et al.
This article has been prepared by Dr Penny Ward FFPM, with input from Professor Tim Higenbottam PFPM, Dr Flic Gabbay FFPM, Dr Bob Holland FFPM, Dr Sue Tansey FFPM and Dr Tahir Saleem MFPM.
It is provided for information and does not constitute advice or represent official FPM views or policy.
How to cite:
Ward, P et al. (2020), ‘COVID-19 Vaccine and Antiviral Drug Development’, Faculty of Pharmaceutical Medicine blog, 08 April 2020. Available at: https://www.fpm.org.uk/blog/covid-19-vaccine-and-antiviral-drug-development/ (Accessed: <date>).
This is the second part of a two-part review.
Read part one
The COVID-19/SARS-CoV-2 Pandemic
Introduction
On 28 January 2020, just weeks after the first official reports of the 2019 novel coronavirus began to emerge from Wuhan, China, the U.S. Health and Human Services called a press conference in Washington, D.C. By this point, SARS-CoV-2 had infected thousands and millions were quarantined in China. No treatment for the virus exists. Said Anthony Fauci, Director National Institute of Allergy and Infectious Disease (NIAID)“We’re looking at the worst scenario that this becomes a bigger outbreak. To stop that outbreak and save lives the world needs a vaccine”.1
Vaccines
Recovery from Disease Confers Immunity to Rechallenge
A recent study in SARS-CoV-2 challenged rhesus macaques has demonstrated that recovery from prior infection is associated with immunity to rechallenge. Thus, a proof of principle for using vaccines to stimulate immunity in humans has been demonstrated2.
Vaccine Technologies
A range of technical approaches to the production of a vaccine are available with advantages and disadvantages to each (Table 1). An RNA vaccine is currently in Phase I trials in healthy volunteers in the USA.
| Vaccine Strategy | Advantages | Disadvantages |
|---|---|---|
| Inactivated vaccines | Easy to prepare; safety; high titre neutralising antibody | Limited immune response in immunosuppressed/elderly |
| Attenuated virus vaccines | Rapid development; induce high immune response (humoral and mucosal) | Pheno/genotypic reversion potential; may cause (mild) disease |
| Subunit vaccines | Safety; high titre antibodies; cellular and humoral response; consistent production | High cost; lower titre antibodies requiring adjuvants or repeated doses; |
| Viral Vector Vaccines | Safety; high response (humoral and cellular) | Requires identification of antigens with neutralizing epitopes from recovered cases |
| DNA vaccines | Simple to design; high safety; high titre antibodies | No currently licensed vaccine in human use; delivery issues; toxicity risk (DNA insertion into human genome; anti DNA antibody stimulation) |
| RNA vaccines | Simple to design; adaptable; induce strong immune response | No currently licensed vaccine in use in humans/animals; stability; scale up limitations; unintended immunogenicity vs vaccine |
Preliminary data investigating antibody production from recovered patients suggests that antibodies to the S protein and the receptor binding domain (RBD) can be neutralizing in vitro3,4. Thus most research has focused on production of vaccines directed against the S protein/subunits and the RBD. Ongoing research is investigating the function of antibodies produced to other viral proteins for both contribution to disease pathology and protection against infection in affected patients/animal models.
Duration of Vaccine Induced Immunity
Several issues remain to be addressed: prior experience following studies of human coronavirus infections causing the common cold have suggested that immunity may be short lived (3 months or less) and individuals may be reinfected within a short period after prior exposure3. However longitudinal studies of antibody mediated immunity from recovered SARS patients suggests that immunity after infection may persist for 1-2 years4. In addition, cellular immunity is also long lasting with prompt elicitation of cytotoxic T cell responses to challenge 1 year post recovery5. As this is a novel virus, persistence of neutralising antibody levels requires follow up of recovered patients over time.
Antibody Dependent Enhancement
The potential for antibody dependent enhancement (ADE) is also a concern: ADE is a result of non neutralizing antibodies facilitating virus entry into cells. This phenomenon has been observed with some other virus infections, most notably Dengue. This phenomenon might be a contributor to the vaccine enhanced disease noted in animal models investigating SARS vaccine response6 and in humans following initial investigations of an RSV vaccine7. For this reason rushing to investigate potential vaccines in humans prior to conducting appropriate animal safety studies is questionable.
Regulatory Considerations for Vaccine Development
A first global regulatory workshop was held on 18 March 2020. It was attended by delegates from 17 countries, representing more than 20 medicines regulatory authorities and included experts from the WHO and the European Commission. The meeting was held to discuss regulatory considerations related to the development of SARS-CoV-2 vaccine candidates and preclinical data required to support proceeding to first-in-human (FIH) clinical trials.
The attendees generally agreed the following:
Quality considerations
- The extent of preclinical data to support proceeding to FIH clinical trials depends on the vaccine construct, the supportive data available for the construct and data from closely related products.
- Opportunities to leverage knowledge accumulated with platform technology should be considered to accelerate the development of a SARS-CoV-2 vaccine manufactured using the same platform.
Toxicology extrapolation for well characterized platform vaccine technologies
- If a platform technology utilized to manufacture a licensed vaccine or other investigational vaccines is well characterized, it is possible to use toxicology data (e.g., data from repeat dose toxicity studies, biodistribution studies) and clinical data accrued with other products using the same platform to support FIH clinical trials for a SARS-CoV-2 vaccine candidate.
- The vaccine manufacturer should provide a rationale supported by data to justify why certain preclinical studies such as toxicity studies would not need to be conducted prior to proceeding to FIH clinical trials.
Safety considerations
- CMC characterization should be adequate to support the safety of the SARS-CoV-2 vaccine construct prior to proceeding to FIH clinical trials.
- For all SARS-CoV-2 vaccine candidates it is necessary to obtain data in animals and to characterize the immune response induced by a SARS-CoV-2 vaccine candidate.
- It is not required to demonstrate the efficacy of the SARS-CoV-2 vaccine candidate in animal challenge models prior to proceeding to FIH clinical trials.
Risk of Vaccine Enhanced Disease
- Participants acknowledged the urgency of proceeding to FIH trials with SARS-CoV-2 vaccine candidate in light of the current pandemic but stressed the importance of risk mitigation strategies so that human subjects enrolled in clinical trials would not need to be exposed to unreasonable risk.
- The potential for vaccine induced disease enhancement is a special circumstance that needs to be evaluated according to available science, which may include the use of relevant animal models currently in development.
- Even though there are limitations in the current knowledge and understanding of risk of enhancement of disease and the value of these models in predicting likelihood of occurrence in humans, studies in animal models are considered important to understand the potential for vaccine induced disease enhancement with SARS-CoV-2 vaccine candidates.
- It needs to be recognized that there is limited availability of non-human primates and requiring such studies with every SARS-CoV-2 vaccine candidate prior to FIH trials is not possible and would significantly delay clinical vaccine development.
- The need to address the potential for vaccine-induced enhanced disease to enable FIH clinical trials with SARS-CoV-2 vaccines should be based on the totality of available data relevant to the particular SARSCoV-2 vaccine candidate including the vaccine construct, the immune response induced by it, e.g., Th1-type skewed immune responses and titers of neutralizing antibodies, and the design of the FIH clinical trial.
Considerations for expedited clinical development
The following points were not supported by consensus but there was general agreement that:
- Some vaccine constructs for which there is adequate support from the knowledge around the immune response elicited, may be allowed to proceed to FIH trials without first completing animal studies to assess the potential for enhanced disease provided adequate risk mitigation strategies are put in place in these FIH trials.
- For some vaccines, preclinical data (e.g. postvaccination challenge data from animal models, immunopathology studies in animal models, etc.) may be required prior to advancing to FIH clinical trials.
- In the event that FIH clinical trials are allowed to proceed in the absence of studies in animals that would address the potential for enhanced disease, such studies are, in general, expected to be conducted in parallel with FIH trials so that these data are available prior to enrolling large numbers of human subjects into Phase 2 and 3 clinical trials.
- Risk mitigation strategies to be considered for FIH clinical trials include enrolment of healthy younger adults, proper informed consent to ensure that subjects are aware of the theoretical risks and careful safety follow up and frequent monitoring.
- Regulators expressed the need for developing mechanisms that would allow sharing of data from animal models and clinical trials to alert the global regulatory community regarding the outcomes of trials in anongoing and timely manner.
Therapeutic approaches
Enhancement of innate immunity
In the absence of a specifically directed antiviral, work is ongoing to investigate the contribution of products enhancing natural innate immunity to viral infections. The innate immune system is the body’s ‘first response’ to infection. Attachment of CoV to the host cell receptor and release of RNA into the cytoplasm stimulates an immediate cellular response, via sensitization of TLR-3, leading to an interferon (IFN) response, which then triggers a cascade of downstream pathways which act together to protect surrounding uninfected cells and limit virus replication in infected cells. These are described in Figure 2.
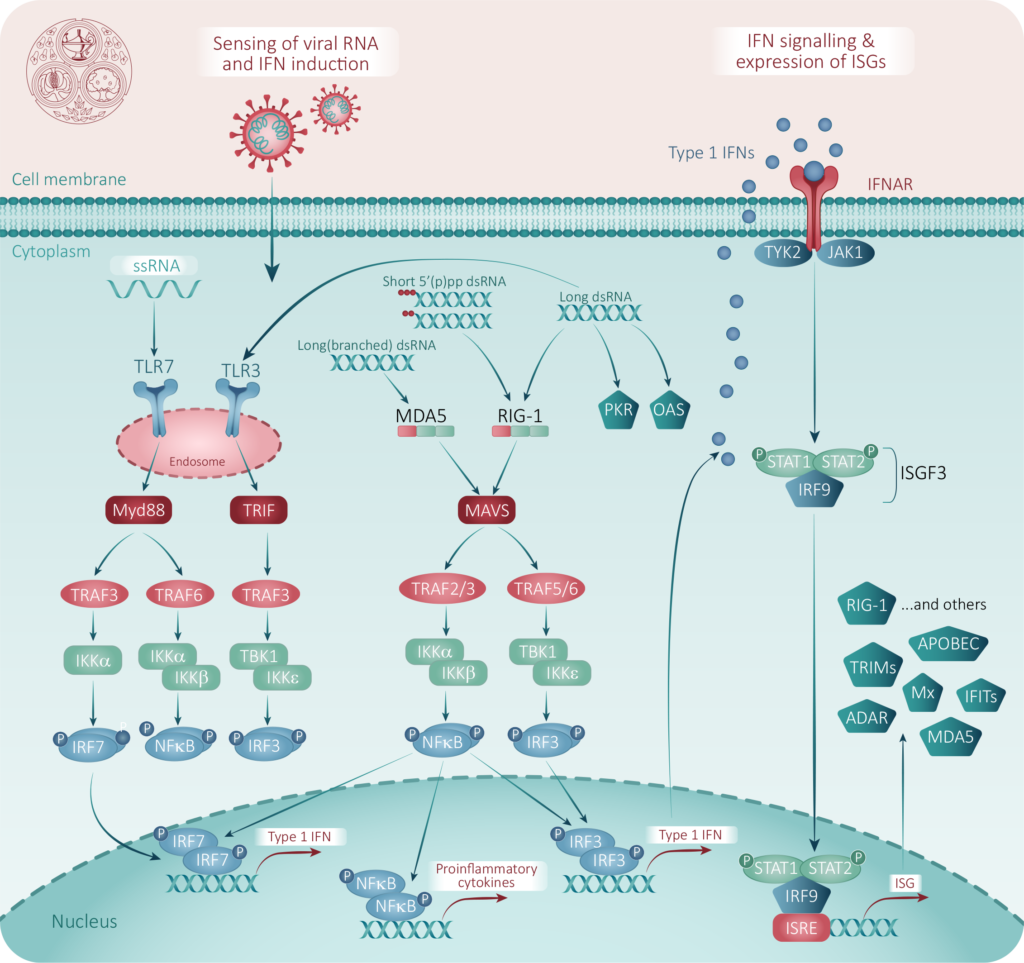
Interferon α and β
High dose IFN treatment has antiviral effects against SARS CoV and MERS CoV in vitro8. Intranasal interferon protects against experimental coronavirus 229E infection9. Systemic interferon has been used to treat patients with SARS and MERS, although clinical effects are unclear 10,11. Multiple clinical trials are underway investigating the potential effect of IFNα in COVID-19.
More recently, administration of inhaled beta interferon (IFN-β) treatment to mild asthmatics with viral colds resulted in a modest, but significant, induction of systemic innate immune responses, which in turn improved the clinical outcome of the cold. In addition, there was a trend towards reduced viral load in sputum observed with treatment suggesting potential for faster clearance of respiratory viruses in this patient group. Morning peak expiratory flow rates were higher among IFN-β recipients, suggesting preservation of airways function associated with antiviral effect12. A clinical trial in patients with COVID-19 is starting shortly in the UK.
Toll-like receptor agonists
PUL-042 is a novel combination of two synthetic molecules (Pam2 and ODN) that are agonists of the Toll-like receptors TLR2/6 and TLR9 respectively. The combination of these two agonists in a molar ratio of 4Pam2:1ODN has been shown to protect against transfection with bacteria, fungi and viral pathogens in a variety of model systems13. Exposure of mice to an aerosolized combination of TLR2/6 and TLR9 ligands induced transient and self-limited neutrophilic inflammation within the lungs that is associated with minimal systemic inflammation or physiologic and behavioral responses, suggesting that it may be feasible to use aerosolized TLR ligands to prevent opportunistic lung infections in immunocompromised patients or in normal subjects to attenuate lung infections with virulent pathogens13. A clinical trial of PUL-042 inhaled solution investigating post exposure prevention of infection/illness in subjects exposed to COVID-19 is about to start.
Antiviral Therapies
Initial analyses of genomic sequences from SARS-CoV-2 (Figure 3 14) indicates that the catalytic sites of the four enzymes that may be suitable targets for antiviral therapy are highly conserved and similar to the sequence of corresponding SARS and MERS CoVs.
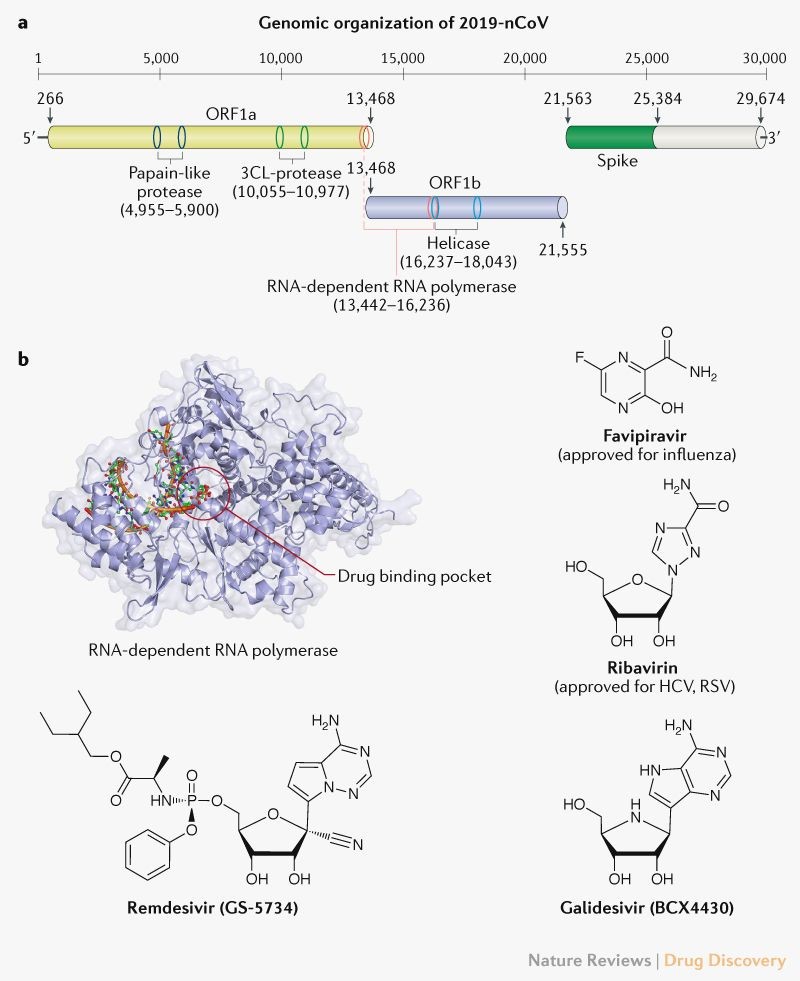
Protein structural analyses suggest that key binding pockets of these viral enzymes are likely to be conserved across the three viruses and it is reasonable to use data from agents already assessed for activity against SARS/MERS CoVs to extrapolate to SARS-CoV-214. Nucleoside analogues can target the rna dependent rna polymerase, while protease inhibitors may be effective against the papain-like and 3Cl-protease enzymes. Lastly, agents targeting the S protein may interfere with virus binding to host cells.
Nucleoside Analogues
The RNA genome of SARS-CoV-2 requires insertion of nucleotides, adenosine, guaninosine, uridine and cytosine. A variety of analogues of these have been developed as anti viral therapies for a range of viral infections including HIV, influenza, hepatitis, and cytomegalovirus.
Favipiravir
The approved nucleoside analogues favipiravir (licensed in Japan as a treatment for influenza) and ribavirin (licensed for the treatment of hepatitis) have been tested against SARS and MERS and shown to have antiviral effects in vitro. Reports from China have suggested that favipiravir had clinical effect in patients with COVID-19 and it has been added to the list of potential therapies there15. Favipiravir is a prodrug and must be metabolised to an actively antiviral form favipiravi- ribofuranosyl-5′-triphosphatephosphate. It has broad spectrum antiviral effects against a range of viruses including influenza, west nile virus, and yellow fever virus. Because it has documented teratogenic effects in animal studies, its use as an in Japan is restricted to emergency use. Favipiravir should not be administered to pregnant women but can otherwise be considered for use in COVID-19 affected patients. In a small open label study comparing favipiravir to lopinavir/ritonavir therapy, favipiravir was associated with significantly increased rate of viral clearance, improvement of pulmonary consolidation on CT scans. The study enrolled subjects aged 16-75 within seven days of symptom onset. Subjects with severe disease (resting respiratory rate >30 breaths/minute, oxygen saturation <93% on room air or requiring ICU care) were excluded. Favipiravir was given at a dose of 1600 mg twice daily on Day 1 and 600 mg twice daily on Days 2–14. LPV/RTV was given orally at a dose of 400 mg/RTV 100 mg twice daily. Both FPV and LPV/RTV were continued until viral clearance was confirmed or until day 14. In addition, all participants received IFN-a1b 60 mg twice daily by aerosol inhalation. Standard care included oxygen inhalation, oral or intravenous rehydration, electrolyte correction, antipyretics, analgesics, and antiemetic drug as necessary.
Ribavirin
Ribavirin is a guanosine analog with in vitro activity against a large number of highly lethal emerging viruses. Mechanistically, ribavirin inhibits RNA synthesis by viral RdRp as well as inhibiting mRNA capping. However, studies demonstrated that while SARS-CoV, MERS-CoV, and HCoV-OC43 were sensitive to ribavirin in vitro, doses that significantly inhibited CoV replication exceeded ribavirin concentrations attainable by typical human regimens16-19. Recently, it was demonstrated that excision of ribavirin nucleoside analogs by conserved coronavirus proofreading mechanisms likely accounted for a greater than expected decrease in in vitro efficacy of ribavirin. Additional in vivo testing of ribavirin in mouse models found limited activity against MA15 SARS-CoV by ribavirin alone and suggested that ribavirin treatment enhanced SARS disease signs20. However, combination treatment of ribavirin and type I Interferons in primate models improved MERS disease signs19. Ribavirin has been given as part of treatment regimens for SARS and MERS patients, but meta-analyses of case studies have found limited (if any) efficacy of ribavirin in treating patients with highly pathogenic coronavirus respiratory syndromes21,22. Despite this past history, studies investigating the combination of ribavirin and interferon α are ongoing in China.
Experimental Nucleoside Analogues
Remdesivir
Remdesivir (GS-5734) is a small molecule adenosine nucleoside analogue that has demonstrated antiviral activity in vitro against several viral families of emerging infectious diseases including Filoviridae, Pneumoviridae, Paramyxoviridae, and Coronaviridae. It was initially studied as a possible treatment for Ebola, but was not particularly effective in comparison to other agents investigated in these outbreaks. Remdesivir has been shown to be effective in animal models of SARS and MERS and is under investigation investigated as a treatment for MERS. Based on activity against MERS-CoV within a larger panel targeting lethal viruses from multiple viral families, additional studies demonstrated that remdesivir decreased viral titers and viral RNA in in vitro models of both SARS-CoV and MERS-CoV infection of HAEs23. Remdesivir has been shown to have inhibitory effects against a diverse range of CoVs including HCoV-NL63 and Mouse Hepatitis Virus (MHV, betacoronavirus group 2a). Importantly, it also inhibited replication of pre-emergent BatCoVs including BatCoVHKU5, BatCoV-HKU3, BatCoV-SHC014, and BatCoV-WIV1. Activity in vivo against CoVs was supported by ameliorated disease signs (weight loss, lung viral titers) in MA15 SARS-CoV infected mice treated prophylactically or therapeutically23. Initial experience with remdesivir in COVID-19 has been encouraging, with rapid reductions in viral load and clinical improvement occurring within 2 days of starting treatment in one patient report24. Remdesivir must be given intravenously with a 200mg loading dose on day 1 and thereafter 100mg daily doses for up to 10 days. Thus this product is most suitable for use in a hospital setting. Multiple trials are ongoing in China, the US and internationally with first results from ongoing studies in China anticipated shortly.
Viral resistance to remdesivir has been shown experimentally in vitro, however the mutations found to conserved motifs in SARS-CoV and MHV resulted in decreased viral fitness in vitro and in vivo, suggesting that a second outbreak of drug resistant CoV infection is unlikely25.
Galidesivir
Galidesivir (BCX4430, Immucillin-A) is an adenosine analog under development by BioCryst Pharmaceuticals. Galidesivir has broad-spectrum antiviral effectiveness against a range of RNA virus families, including bunyaviruses, arenaviruses, paramyxoviruses, coronaviruses (Including SARS and MERS), flaviviruses and phleboviruses26. A Phase 1 clinical safety and pharmacokinetics study in healthy subjects has been completed. As with remdesivir, this product is administered intravenously.
Other Agents
Work using computer modelling of drug binding to active targets on the coronavirus genome has been going on to identify other agents which might have activity against SARS CoV2 targets14,27: this work has identified possible binding activity for a range of other nucleosides including acyclovir, ganciclovir, entecavir, penciclovir, lamivudine and abacavir but no specific studies have been reported documenting antiviral effects against SARS-CoV-2 in vitro or in vivo at this time. In addition a range of non nucleoside reverse transcriptase inhibitors of HIV-1 including efavirenz and etravirine might have some activity, but this has yet to be confirmed.
Viral Protease Inhibitors
Lopinavir/ritonavir
Lopinavir/ritonavir, a protease inhibitor used to treat HIV/AIDS, was found to block COVID-19 infection in vitro at low-micromolar concentration, with a half-maximal effective concentration (EC50) of 8.5 μM. In contrast in vitro inhibitory concentrations vs HIV are in the low nM range (~6nM). Depending on cell type used, the EC50 of lopinavir in vitro for SARS-CoV has ranged from 4.0 to 10.7 μg per milliliter although some studies reported that lopinavir was inactive or that higher concentrations (25 μg /mL) were required for inhibition. For MERS-CoV, the EC50 values have ranged from 5 to approximately 7 μg/mL). Both the mean peak (9.6 μg/mL) and trough (5.5 μg/mL) serum concentrations of lopinavir in adults just approach these concentrations. In general, for anti-infectives, it is desirable to exceed the EC90 concentrations or to sustain concentrations above the EC90 in order to have a reasonable chance of effect. In the first randomized trial investigating the impact of Lopinavir/ritonavir on hospitalized patients with COVID-19, no overall effect was observed in the ITT population28. Of note treatment in this study began >10 days post symptom onset, which would be later than desired given the shape of viral kinetics in COVID-19. Further trials with this agent should seek to investigate higher doses and earlier start of treatment.
Other protease inhibitors
A range of other agents targeting the proteases of HIV-1 or hepatitis viruses have been investigated in computer modelling studies. Marketed products with some predicted binding to coronavirus enzymes include nelfinavir, darunavir, atazanavir, grazoprevir, telaprevir and indinavir14. At this time there is no specific in vitro or in vivo SARS CoV inhibitory data supporting use of these agents in clinical studies. Janssen has recently taken the unusual step of advising against use of darunavir in clinical studies until preclinical evidence of activity has been generated29. Experimental agents which may have some activity based on computer modelling studies includes asunaprevir, which is under investigation as a possible therapy for hepatitis C.
Anti-malarials
Chloroquine
In vitro, chloroquine has antiviral activity against diverse RNA viruses including rabies, poliovirus, HIV, hepatitis A virus, hepatitis C virus, influenza A and B viruses, Chikungunya virus, Dengue virus, Zika virus, Lassa virus, Hendra and Nipah viruses, Crimean–Congo hemorrhagic fever virus and Ebola virus, as well as various DNA viruses. Chloroquine is known to block virus infection by increasing endosomal pH required for virus/cell fusion, as well as interfering with the glycosylation of cellular receptors of SARS-CoV. In a time-of-addition assay chloroquine functioned at both entry, and at post-entry stages of SARS-CoV-2 infection in Vero E6 cells. In addition to its antiviral activity, chloroquine has an immune-modulating activity, which may synergistically enhance its antiviral effect in vivo30. Chloroquine is widely distributed in the body, including lung tissue, after oral administration31. The EC90 value of chloroquine against SARS CoV2 in Vero E6 cells was 6.90 μM, which is clinically achievable and demonstrated in the plasma of rheumatoid arthritis patients following a 500mg oral dose. Chloroquine is an inexpensive product that has been used for more than 70 years. Although its side effect profile includes potential for retinopathy and cardiac dysfunction, these effects occur most frequently with prolonged rather than short term use. Chloroquine has been used for the treatment of COVID-19 patients in China and was reported at a news conference to have had beneficial effect, but published data is awaited from the many ongoing studies are awaited.
Hydroxychloroquine
Hydroxychloroquine is an analog of chloroquine which is less likely to be affected by potential for drug-drug interactions. In the 2003 SARS outbreak, hydroxychloroquine was reported to have anti-SARS-CoV activity in vitro, suggesting that hydroxychloroquine may be a potential pharmacological agent for the treatment of COVID-19 infection. Yao and colleagues explored the in vitro potency of hydroxychloroquine vs SARS-CoV-231. Both chloroquine and hydroxychloroquine were found to decrease the viral replication in a concentration dependent manner with EC50 values for chloroquine of 23.90 and 5.47 μM at 24 and 48 hours, respectively and for hydroxychloroquine 6.14 and 0.72 μM at 24 and 48 hours respectively. Based on these data the authors used a PBPK model to predict the dose regimen of hydroxychloroquine which would provide antiviral drug levels throughout the dosing period; the model suggests a loading dose of 400mg given twice on day 1 followed by 200mg daily for the next 5 days may be adequate. This deserves investigation in clinical trials.
Treatment of COVID-19 induced Cytokine Storm
Investigation of subjects with severe COVID-19 disease suggests that an exaggerated immune response to infection may contribute to worsening disease and the eventual development of ARDS and multiorgan dysfunction. A variety of cytokines contribute to this syndrome including IL-1, IL-6 and IFN-γ32. Antagonist antibodies inhibiting IL-1 (anakinra, rilonacept and canakinumab), IL-6 (tocilizumab, sarilumab) and IFN-γ (emapalumab) have been approved for the treatment of inflammatory disorders mediated by these cytokines and may be useful in downregulating the cytokine storm associated with progression to ARDS in some COVID-19 patients. Tocilizumab has been used for the management of severe COVID-19 infection in China and is anecdotally reported to result in rapid defervescence and recovery of lung abnormalities in treated patients. Several trials clinical trials are in progress and are being planned to explore the efficacy of this product, and the competing anti IL-6 antibody sarilumab. Emapalumab is an antagonistic antibody to IFN-γ approved in the US for the treatment of hemophagocytic lymphohistiocytosis (HLH). A trial of this agent in the management of COVID-19 is being conducted in Italy.
JAK-STAT Inhibitors
Benevolent AI has reported on a computer based assessment of agents which might inhibit clathrin mediated viral endocytosis and thereby inhibit viral infection of cells. The drug targets are members of the numb-associated kinase (NAK) family—including AAK1 and GAK—the inhibition of which has been shown to reduce viral infection in vitro. Baricitinib was identified as a NAK inhibitor, with a particularly high affinity for AAK1, a pivotal regulator of clathrin mediated endocytosis and may be of assistance in SARS-CoV-2 infection. In addition, JAK–STAT signalling inhibitors are likely to be effective against the consequences of the elevated levels of cytokines (including interferon-γ) associated with later complications of infection including ARDS. At therapeutic doses used for the treatment of patients with rheumatoid arthritis, the free plasma concentrations of baricitinib are predicted to be sufficient to inhibit AAK1, and potentially GAK, in cell-based assays, whereas other JAK inhibitors are unlikely to be associated with this effect at tolerated doses used for rheumatoid arthritis treatment33. Clinical trials with baracitinib are being conducted in the US and China as monotherapy and in combination with other experimental antiviral regimes.
Inhibitors of Complement Activation
Complement activation is an important part of the innate immune system and can be initiated both by bacterial and viral infections. Complement activation is associated with pathology in several rare diseases including paroxysmal nocturnal haemoglobinuria (PNH) and thrombotic microangiopathy associated with human stem cell transplant. The complement inhibitor eculizumab34 is an effective treatment for PNH. Trials with eculizumab have begun in patients with cytokine storm symptoms associated with COVID-19 pneumonia. Another recent entrant antibody treatment, IFRX-1, is in trials in China. A recombinant protein inhibitor of C5a and LTB4, nomacopan, which has shown preliminary efficacy in these disorders is also being investigated. These therapies may not only calm the exuberant underlying inflammation but also the reverse intravascular coagulation (indicated with high D dimer level and low platelet counts) which has been noted in COVID-19 patients that fail to survive.
Conclusions
The emergence and rapid spread of SARS-CoV-2 represents a major emerging health threat to countries and populations around the world. Despite the rapidity of progress of the disease, the pharmaceutical industry has stepped up research to understand the disease pathogenesis and to convert that understanding to the rapid scale up and production of therapeutic and prophylactic approaches to protect exposed populations. In addition, based on prior understanding gained from multiple prior investigations of vaccine and antiviral agents, regulators are considering reasonable amendments to regulatory requirements to enable fast tracking of appropriate candidates into clinical studies. The world awaits the outcomes of these combined efforts.
References
- https://www.nature.com/articles/d42473-020-00032-z
- Bao LL et al. Reinfection could not occur in SARS-CoV-2 infected rhesus macaques. bioRxiv preprint doi: https://doi.org/10.1101/2020.03.13.990226
- Callow, K.A., Parry, H.F., Sergeant, M., and Tyrrell, D.A. (1990). The time course of the immune response to experimental coronavirus infection of man. Epidemiol Infect 105, 435-446.
- Mo H et al. Longitudinal profile of antibodies against SARS-coronavirus in SARS patients and their clinical significance. Respirology 2006; 11 : 49–53
- Li C.K., Xu X. (2010) Host Immune Responses to SARS Coronavirus in Humans. In: Lal S. (eds) Molecular Biology of the SARS-Coronavirus. Springer, Berlin, Heidelberg.
- Wang SF et al. Antibody-dependent SARS coronavirus infection is mediated by antibodies against spike proteins. Biochem Biophys Res Commun 2014; 451: 208-214
- Kim H W et al. Respiratory Syncytial Virus Disease in Infants Despite Prior Administration of Antigenic Inactivated Vaccine. Am J Epidemiol 1969; 89: 422–434
- Cinatl J et al. Treatment of SARS with human interferons. Lancet 2003; 362: 293-294
- Higgins, P.G. et al. Intranasal interferon as protection against experimental respiratory coronavirus infection in volunteers. Antimicrobial Agents and Chemotherapy 1983; 24: 713-715.
- Loutfy MR, et al. Interferon alfacon-1 plus corticosteroids in severe acute respiratory syndrome: a preliminary study. JAMA: the journal of the American Medical Association. 2003; 290:3222–3228
- Zhao Z et al. Description and clinical treatment of an early outbreak of severe acute respiratory syndrome (SARS) in Guangzhou, PR China. J Med Microbiol 2003; 52: 715-20.
- Djukanovic R et al. The Effect of Inhaled IFN-β on Worsening of Asthma Symptoms Caused by Viral Infections. A Randomized Trial. Am J Respir Crit Care 2013; 190: 145–154
- Alfaro VY et al. Safety, tolerability, and biomarkers of the treatment of mice with aerosolized Toll-like receptor ligands. Frontiers in Pharmacology 2014; https://doi.org/10.3389/fphar. 2014.00008
- Li G and Le Clercq E. Therapeutic options for the 2019 novel coronavirus (2019-nCoV). Nature Reviews Drug Discovery 2020; 19: 149-150
- Cai Q et al. Experimental Treatment with Favipiravir for COVID-19: An Open-Label Control Study. Engineering 2020. https://doi.org/10.1016/j.eng.2020.03.007
- Shen L et al. Safe and sensitive antiviral screening platform based on recombinant human coronavirus OC43 expressing the luciferase reporter gene. Antimicrob Agents Chemother. 2016; 60: 5492–5503.
- Hart BJ et al. Interferon-β and mycophenolic acid are potent inhibitors of Middle East respiratory syndrome coronavirus in cell-based assays. J Gen Virol. 2014; 95: 571–577.
- Ströher U et al. Severe acute respiratory syndrome-related coronavirus is inhibited by interferon-α. J Infect Dis. 2004; 189: 1164–1167.
- Falzarano D et al. Inhibition of novel β coronavirus replication by a combination of interferon-α2b and ribavirin. Sci Rep. 2013; 3: 1686.
- Barnard DL et al. Enhancement of the infectivity of SARS-CoV in BALB/c mice by IMP dehydrogenase inhibitors, including ribavirin. Antiviral Res. 2006;71: 53–63.
- Stockman LJ et al. SARS: systematic review of treatment effects. PLoS Med. 2006; 3: e343.
- Morra ME et al. Clinical outcomes of current medical approaches for Middle East respiratory syndrome: a systematic review and meta-analysis. Rev Med Virol. 2018; 28: e1977.
- Sheahan TP et al. Broad-spectrum antiviral GS-5734 inhibits both epidemic and zoonotic coronaviruses. Sci Transl Med. 2017; 9 (396). DOI:10.1126/scitranslmed.aal3653
- Holshue ML et al. First Case of 2019 Novel Coronavirus in the United States. N Engl J Med 2020; 382:929-936. DOI: 10.1056/NEJMoa2001191
- Agostini ML et al. Coronavirus susceptibility to the antiviral remdesivir (GS-5734) is mediated by the viral polymerase and the proofreading exoribonuclease. mBio. 2018; 9. DOI:10.1128/ mBio.00221-18
- Zumla A et al. Coronaviruses — drug discovery and therapeutic options. Nat. Rev. Drug Discov 2016; 15: 327–347
- Beck BR et al. Predicting commercially available antiviral drugs that may act on the novel coronavirus (2019-nCoV), Wuhan, China through a drug-target interaction deep learning model. bioRxiv preprint doi: https://doi.org/10.1101/2020.01.31.929547.
- Cao B et al. A Trial of Lopinavir–Ritonavir in Adults Hospitalized with Severe Covid-19. NEJM 2020.
- https://www.janssen.com/uk/sars-cov-2-treatment accessed 30 March 2020.
- Devaux CA et al. New insights on the antiviral effects of chloroquine against coronavirus: what to expect for COVID-19? International Journal of Antimicrobial Agents 2020; doi:
https://doi.org/10.1016/j.ijantimicag.2020.105938 - Yao X et al. In Vitro Antiviral Activity and Projection of Optimized Dosing Design of Hydroxychloroquine for the T reatment of Severe Acute Respiratory Syndrome Coronavirus 2 ( SARS-CoV-2). Clin Infect Dis. 2020 Mar 9. pii: ciaa237. doi: 10.1093/cid/ciaa237
- Conti P et al. Induction of pro-inflammatory cytokines (IL-1 and IL-6) and lung inflammation by Coronavirus-19: anti-inflammatory strategies. J Biol Regul Homeost Agents 2020; 34: doi 10.23812
- Richardson P et al. Baricitinib as potential treatment for 2019-nCoV acute respiratory disease. Lancet 2020. 24 February. https://doi.org/10.1016/S0140-6736(20)30304-4
- Parker, C., Kar, S. & Kirkpatrick, P. Eculizumab. Nat Rev Drug Discov 2007; 6: 515–516.